Введение
Биоинформатика и большие объемы данных
Наука биоинформатика занимается поиском инструментов и выполнением анализов, которые позволяют лучше понять механизмы молекулярных взаимодействий, приведшие к возникновению жизни на планете Земля, в большей степени благодаря анализу и упорядочиванию геномной и протеомной информации. По мере сбора огромных объемов геномной информации, включающей как геномные последовательности, так и выраженные последовательности генов, критичными становятся более эффективные, точные и специфичные методики выполнения анализа этой информации.
При формировании последовательностей ДНК химические и механические процессы по большому счету позволяют "оцифровать" информацию, присутствующую в ДНК и РНК. Эти последовательности записываются с помощью алфавита , в котором каждому нуклеотиду соответствует одна буква. Для определения того, как эти данные последовательностей структурируются с целью формирования строительных блоков большего размера, а также того, как они связаны с другими данными последовательностей, выполняются различные анализы. Эти операции лежат в основе процесса изучения биологической эволюции и развития живых организмов, генетики и, все чаще, медицины.
Данные цепочек нуклеотидов, получаемые в процессе исследования их последовательностей, представляются в форме строк, состоящих из символов, известных, как чтения . (Способ применения термина "чтение" (read) в области биоинформатики вызывает неприятное противоречие со способом применения этого термина в областях компьютерных наук и разработки программного обеспечения. Это особенно актуально по причине того, что производительность чтений может быть оптимизирована, о чем мы еще поговорим. Для разрешения данного неприятного противоречия мы будем называть последовательности из геномов "геномными чтениями" (genomic reads).) Для анализа широкомасштабных структур и процессов необходимо осуществить объединение множества геномных чтений. Это объединение сложнее объединения мозаики по той причине, что результирующая картинка обычно неизвестна априори и элементы могут перекрываться (что обычно и происходит). Дополнительное усложнение заключается в том, что не все геномные чтения выполняются достаточно точно, поэтому они могут содержать множество различных ошибок, таких, как избыточные или недостающие символы, а также некорректные символы, обозначающие нуклеотиды. Хотя дополнительные чтения и могут помочь помочь в сборке или проверке элементов мозаики, они также могут содержать ошибки ввиду не идеальной точности всех существующих методик изучения последовательностей. Частота появления ошибочных геномных чтений возрастает с возрастанием объема данных, что еще более осложняет сбор данных.
По мере совершенствования технологии изучения последовательностей, объем генерируемых данных последовательностей начал превосходить возможности аппаратного обеспечения компьютеров, которые выполняли анализ таких данных в соответствии с устоявшимися методиками. (Большая часть внедренных технологий изучения последовательностей генерирует огромные количества геномных чтений, обычно от десятков миллионов до миллиардов, причем каждое из чтений является последовательностью, содержащей описания от 50 до 100 нуклеотидов.) Эта тенденция продолжает развиваться и является примером проблемы обработки больших объемов данных ( Big Data problem ) [1], актуальной для участников сообществ пользователей вычислительных систем высокой производительности (HPC), систем анализа и исследователей методов обработки информации. По причине того, что сдерживающим фактором начали становиться возможности аппаратного обеспечения, все большее внимание начало уделяться способам решения возникшей проблемы на уровне программного обеспечения. В данной главе мы представим одно из таких программных решений, а также опишем наш метод его применения и масштабирования для обработки терабайт данных.
Наше исследование было направлено на реализацию эффективной системы предварительной обработки данных , в рамках которой с помощью различных фильтров и методов хранения данных могли осуществляться усечения, удаления и сохранения данных геномных чтений с целью упрощения последующих анализов. Преимущество этого подхода заключается в ограничении количества изменений, которые должны проводиться при последующих анализах, данные геномных чтений для которых в общем случае могут передаваться напрямую.
В данной главе мы представим наше программное решение и опишем наши способы его настройки и масштабирования для эффективной обработки постоянно увеличивающихся значительных объемов данных.
Чем является программное обеспечение khmer?
Khmer - это наш набор программных инструментов для предварительной обработки больших объемов данных геномных последовательностей перед осуществлением их анализа с помощью стандартных инструментов из области биоинформатики [2] - он не имеет никакого отношения к этнической группе из юго-восточной Азии. Название этого набора инструментов возникло благодаря свободной ассоциации термина k-mer (k-мер): в ходе предварительной обработки генетические последовательности разделяются на перекрывающиеся подстроки заданной длины k . Так как цепочки из множества молекул обычно называются полимерами (polymers), цепочки из заданного количества молекул называются k-мерами (k-mers), причем в этом случае каждая подстрока представляет одну такую цепочку. Следует помнить, что для каждого геномного чтения количество k-меров будет равно количеству нуклеотидов в последовательности за вычетом значения k плюс один. Таким образом, практически каждое геномное чтение будет разделяться на множество перекрывающихся k-меров.
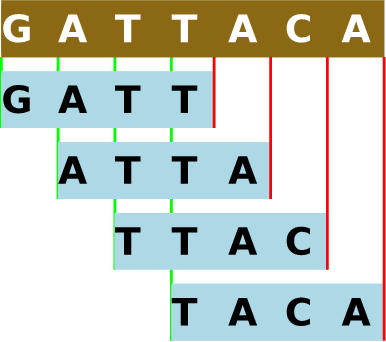
Рисунок 12.1 - Разделение геномной последовательности на 4-меры. В khmer для прямой последовательности и обратного дополнения каждого k-мера используется одно и то же хэшированное значение из-за того, что ДНК состоит из двух цепочек. Подробнее об этом будет сказано в разделе "Направления будущего развития".
Так как мы хотим рассказать вам о том, как мы тестировали и настраивали это программное обеспечение с открытым исходным кодом, мы пропустим большую часть теоретических положений, влияющих на принцип его работы. Достаточно сказать о том, что подсчет количества k-меров является основным этапом большей части операцией. Для упрощения подсчета большого количества k-меров используется структура данных под названием Bloom filter [3]. Вооружившись информацией о количестве k-меров, мы можем исключить избыточные данные из процесса последующей обработки, известного, как "цифровая нормализация". Мы также можем трактовать редко встречаемые данные последовательностей как возможные ошибки и исключать их из последующего исследования для сокращения количества ошибок. Эти процессы нормализации и исключения позволяют в значительной степени сократить объем необработанных данных последовательностей, необходимых для последующего анализа, при условии сохранения большей части интересующей информации.

Рисунок 12.2 - Представление набора программного обеспечения khmer в виде уровней
Основная часть программного обеспечения разработана с использованием языка программирования C++. Эта основная часть состоит из поставщика данных (data pump - компонента, который перемещает данные из сетевого хранилища в физическую оперативную память), систем разбора геномных чтений в нескольких стандартных форматах, а также нескольких систем подсчета k-меров. Интерфейс программирования приложений (application programming interface - API) реализован поверх основной части программных компонентов. Этот API может, конечно же, использоваться из программ на языке C++, как мы делаем при разработке наших тестов, но он также является основой для реализации обертки на языке программирования Python. Пакет для языка Python строится на основе описанной обертки для Python. Множество сценариев на языке программирования Python распространяется вместе с рассматриваемым пакетом программного обеспечения. Таким образом, набор программного обеспечения khmer в совокупности является комбинацией основных программных компонентов, разработанных с использованием языка C++ для повышения скорости работы, высокоуровневых интерфейсов, раскрываемых с использованием языка программирования Python для простоты манипуляций, а также ассортимента инструментов в виде сценариев, которые предоставляют удобные способы выполнения различных задач из области биоинформатики.
Набор программного обеспечения khmer поддерживает возможность выполнения очередей операций во множестве фаз, на каждой из которых могут использоваться отдельные входные и выходные данные. Например, он может принимать набор данных геномных чтений, подсчитывать количество k-меров в них, после чего, в случае необходимости, сохранять хэш-таблицы структуры данных Bloom filter для последующего использования. Позднее он может использовать сохраненные хэш-таблицы для выполнения фильтрации редких k-меров для нового набора геномных чтений с сохранением отфильтрованных данных. Такая гибкость и возможность повторного использования ранее выведенных данных, а также возможность принятия решения о том, какие данные следует сохранять, позволяют пользователю адаптировать процедуру к специфике его или ее потребностей и ограничениям хранилища данных.
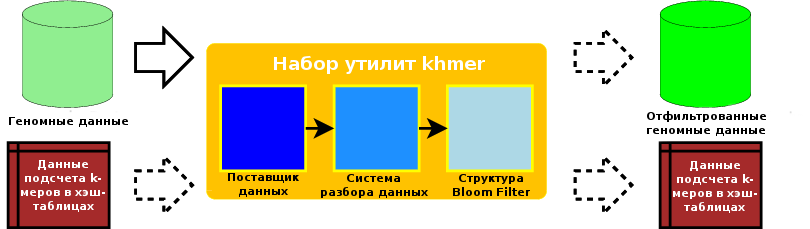
Рисунок 12.3 - Потоки данных в рамках набора программного обеспечения khmer
Огромные объемы данных (возможно, терабайты) должны быть перемещены с диска в память силами программного обеспечения. Использование эффективного поставщика данных необходимо, так как пропускная способность канала от хранилища данных к центральному процессору может быть в три или даже в четыре раза меньше, чем пропускная способность канала для передачи данных из физической оперативной памяти к центральному процессору. Для отдельных типов файлов данных должен использоваться декомпрессор. В любом случае, система разбора должна работать с результирующими данными эффективно. Задача разбора данных заключается в обработке строк переменной длины, при этом также должно учитываться наличие некорректных геномных чтений и сохранятся определенные фрагменты биологической информации, которые могут быть использованы в ходе последующего объединения данных, такого, как объединение концов фрагментов последовательностей. Каждое геномное чтение разделяется на набор перекрывающихся k-меров, а каждый k-мер регистрируется или сравнивается с помощью структуры Bloom filter. В том случае, если ранее данные структуры Bloom filter были обновлены или использованы для сравнения, они должны быть загружены из хранилища. В том же случае, когда структура Bloom filter создавалась для последующего использования или обновлялась, эти данные должны сохраняться в хранилище.
Поставщик данных постоянно осуществляет операции последовательного доступа к данным файлов и теоретически может получить запрос на одновременное чтение нескольких фрагментов данных большого размера. Зная об этом, мы можем сформулировать следующие вопросы:
- В полной ли мере мы используем тот факт, что осуществляется последовательный доступ к данным?
- Достаточно ли данных размещается на страницах памяти для минимизации задержки при доступе к этим данным?
- Может ли асинхронный ввод использоваться вместо синхронного?
- Можем ли мы эффективно преодолеть системные кэши с целью сокращения операций копирования данных из буфера в буфер в памяти?
- Предоставляет ли поставщик данных доступ к этим данным системе разбора таким способом, при котором не используется излишняя логика контроля доступа и принятия решений?
Эффективность работы системы разбора данных очень важна, так как данные предоставляются в достаточно свободном строковом формате и должны преобразовываться во внутреннее представление перед любыми последующими операциями обработки. Так как каждая отдельная запись имеет относительно малый размер (100 - 200 байт), но при этом количество таких записей исчисляется значениями от миллионов до миллиардов, мы в большей степени сфокусировали свое внимание на оптимизации системы разбора записей. Система разбора записей в своей основе представлена циклом, в котором поток данных разделяется на геномные чтения, сохраняемые в записях, причем в процессе обработки данных осуществляются некоторые начальные проверки корректности этих данных.
Некоторые соображения относительно эффективности системы разбора данных:
- Минимизировали ли мы количество операций, в ходе которых система разбора осуществляет доступ к данным в памяти?
- Минимизировали ли мы количество операций копирования данных из буфера в буфер при извлечении геномных чтений из потока данных в ходе его разбора данных?
- Минимизировали ли мы количество вызовов функций внутри цикла разбора данных?
- Система разбора данных должна обрабатывать некорректные данные, включая неоднозначные обозначения, слишком короткие геномные чтения и символы разного регистра. Выполнена ли проверка корректности последовательности ДНК так эффективно, как это возможно?
При рассмотрении механизма итерации между k-мерами в геномном чтении и их хэширования, мы можем задаться следующими вопросами:
- Может ли механизм итерации между k-мерами быть оптимизирован в плане как потребления памяти, так и скорости работы?
- Могут ли функции хэширования, предназначенные для работы со структурой Bloom Filter быть оптимизированы каким-либо образом?
- Минимизировали ли мы количество обращений механизма хэширования к данным в памяти?
- Можем ли мы повысить количество хэшей при конвейерной обработке для эксплуатации методики горячего кэша?